


Therapeutic proteins: facing the challenges of glycobiology
-
Copyright
© 2014 PRO MEDICINA Foundation, Published by PRO MEDICINA Foundation
User License
The journal provides published content under the terms of the Creative Commons 4.0 Attribution-International Non-Commercial Use (CC BY-NC 4.0) license.
Authors
| Name | Affiliation | |
|---|---|---|
Simone Albrecht |
NIBRT, Dublin, Ireland |
|
Mark Hilliard |
NIBRT, Dublin, Ireland |
|
Pauline Rudd |
NIBRT, Dublin, Ireland |
The biologics sector is experiencing tremendous growth worldwide and is fuelled by the launch of a vast product range targeting mainly cancer, autoimmune diseases and hormone/enzyme disorders. However, biologics are one of the most expensive therapeutics to produce, due to both their inherent structural complexity and variability which challenges their manufacturing process and requires a thorough understanding of the product characteristics. More than one third of therapeutic proteins are glycoproteins such as monoclonal antibodies, cytokines, hormones, growth factors, clotting factors, enzymes as well as fusion proteins. Glycosylation is a major post-translational modification (PTM) and a tightly regulated critical quality parameter in the production of therapeutic proteins. This review includes a comprehensive overview on critical glycosylation and production parameters of different classes of therapeutic glycoproteins. It highlights the significance of protein glycosylation in product efficacy, stability and immunogenicity as well as in the development and regulation of follow-on biosimilar products which are set to vastly transform the biologics market in the coming decade.
Introduction
Since the introduction of recombinant DNA technologies in the 1980’s the biologics market has experienced rapid growth including the successful launch of a vast variety of products such as cytokines, hormones, enzymes, fusion proteins and monoclonal antibodies (mAbs). By 2018 biologics are forecasted to account for one quarter of all drug expenses worldwide [1]. However, despite their growing demand, biologics are one of the most expensive pharmaceutical drugs. Biopharmaceuticals are structurally complex molecules and more than one third of approved biopharmaceuticals are glycoproteins. Glycosylation is the most abundant and most structurally diverse post translational modification (PTM). Other features include amidation, sulfation, hydroxylation and carboxylation in proteins [2]. Protein glycosylation in the ER and Golgi results in a complex set of N- and/or O-glycans conferring significant micro- and macro-heterogeneity to the molecule [3]. Representative N- and O-glycans are represented in Figure 1.

Figure 1. Representative N- and O-glycan structures. Complex-type N-glycans (1) are the most abundantly present N-glycan type on therapeutic glycoproteins. These N-glycans can be a-galactosylated [G0] (i), mono-galactosylated [G1] (ii), di-galactosylated [G2] (iii), tri-galactosylated [G3] (not shown) and tetra-galactosylated [G4] (iv), core-fucosylated (iii, iv) and sialylated (ii, iv) and can carry up to four antennas (iv). N-glycans of high-mannose type (2) and hybrid type (3) are generally less frequent. O-glycans on therapeutic proteins are mainly of the core 1 type (4). O-glycans can be additionally extended by Gal-GlcNAc repeats and modified by sialylation, fucosylation, sulphatation, methylation, or acetylation.Glycans are represented using the Oxford symbol nomenclature [113]
N-glycans are typically composed of a core pentasaccharide unit (Man3GlcNAc2) which is linked via a chitobiose (GlcNAc2) to an asparagine (Asn) residue on the Asn-X-Ser/Thr-protein (serine/threonine) consensus sequence. Pharmaceutical glycoproteins produced in mammalian cell expression systems predominantly carry multi-antennary complex-type N-glycans which can be core-fucosylated and sialylated [4]. O-glycans on pharmaceutical glycoproteins are mainly of the core 1 type [Gal(β1,3) GalNAc] and are attached to the glycoprotein via a Ser/Thr residue [5,6]. The glycosylations macro- and micro-heterogeneity of proteins can significantly influence product efficacy and immunogenicity and is highly dependent on cell culture and manufacturing conditions and thus, the glycosylation of biopharmaceuticals presents an important quality and safety parameter. The manufacturing process of biologics is therefore challenging, expensive and time-consuming and can take up to 15 years from the pre-clinical phase until final market approval. Regulatory frameworks require the demonstration of proper glycosylation within acceptable variation limits and require the integration of strict and detailed quality control parameters into the manufacturing process [7,8].
The recent approval of the first biosimilar antibodies in Europe represents a major landmark in the young history of biologic therapeutics [9]. Additionally, more than 70 mAbs are in pre-clinical development [10] which are expected to have comprehensive market implications such as a significant price reduction in the range of 20-30% [11,12]. To date, a total of 18 biosimilars within the product classes of human growth hormone, granulocyte colony-stimulating factor (GCSF), erythropoietin (EPO) and TNF-inhibitor have already been approved for use in the EU [13]. Biosimilars can be defined as follow-on products of an innovator biopharmaceutical for which the patent has expired. The approval of biosimilars follows an abbreviated regulatory pathway but comprehensive comparability studies are required as laid out in guidelines issued by the FDA and EMA [14,15]. Due to the complex nature of biologics and the manifold influences during the production process an absolute similarity cannot be reached. Therefore an extensive dataset derived from pharmacokinetic bioequivalence testing and biophysical characterization is required in order to guarantee safety and efficacy of the biosimilar.
This review includes a comprehensive introduction to the different classes of therapeutic glycoproteins and includes details of the associated critical glycosylation parameters and critical production parameters, such as cell lines and culture conditions. Through our own research activities in the development of high-throughput glycotechnology for quantitative, detailed structural analysis of protein N- and O-glycosylation we have gained a solid insight into the complexity of post-translation glycosylation and the concomitant challenges of their analytical characterization. We also relay interesting case-studies on the glycosylation variability of therapeutic proteins which show that altered glycosylation does not automatically implicate changes in product quality and regulatory rejection. The understanding of the structure-function relationship is therefore a key requirement in the production of biologics.
Therapeutic proteins classes
Pharmaceutical glycoproteins can be sub-divided into different product classes, the largest and best selling of which are the monoclonal antibodies. Other important biologics product classes include glyco-engineered Fc IgG fusion proteins, cytokines, growth factors, hormones and enzymes.
The glycosylation heterogeneity of proteins largely determines their therapeutic effector function and should closely resemble human protein glycosylation. In order to obtain a human-like glycosylation pattern most therapeutic glycoproteins are produced in eukaryotic cell lines, such as Chinese Hamster Ovary (CHO), myeloma (NS0) or hybridoma (SP2/0) [16].
Monoclonal antibodies (mAbs)
Therapeutic mAbs are recombinant immunoglobulins (IgG) mainly of the IgG1 subtype which have a monovalent epitope affinity to specific antigens. Antibodies utilised in the treatment of cancer and autoimmune diseases form the main therapeutic areas and constitute about 80% of the total antibody sales in the US [17]. A comprehensive overview on mAbs and their therapeutic use is given in Table 1.
Table 1. Commercial EMA- and FDA-approved glycosylated monoclonal antibodies for therapeutic use [114]
| Target | applications (examples) | Proprietary (commercial) name | cell line | |
| Anti-inflammatory | TNFα | Rheumatoid arthritisCrohn’s diseaseUlcerative colitis | Adalimumab (Humira®) | CHO |
| Golimumab (Simpoli®) | SP2/0 | |||
| Infliximab (Remicade®) | SP2/0 | |||
| BLys | Systemic lupus erythematosus | Belimumab (Benlysta®) | NS0 | |
| CD3 | Transplant rejection | Muromonab (Orthoclone-OKT3®) | Hybridoma | |
| Basiliximab (Simulect®) | NSO | |||
| IL1β | Cryopyrin-associated periodic syndroms | Canakinumab (Ilaris®) | SP2/0 | |
| IL6R | Rheumatoid arthritisJuvenile idiopathic arthritis | Tocilizumab (Actemra®) | CHO | |
| IL12/IL23 | Plaque psoriasis | Ustekinumab (Stelara®) | SP2/0 | |
| α4-integrin | Multiple sclerosisRheumatoid arthritis | Natalizumab (Tysabri®) | NS/0 | |
| IgE | Asthma | Omalizumab (Xolair®) | CHO | |
| Anti-cancer | CD20 | Non-Hodgkin’S lymphomaChronic lymphocytic leukemiaRheumatoid arthritis | Rituximab (Rituxan®, MabThera®) | CHO |
| Obinutuzumab* (Gazyva®) | CHO | |||
| Ofatumumab (Arzerra®) | NS0 | |||
| CD52 | Leukemia | Alemtuzumab (Campath®, Mabcampath®) | CHO | |
| CTLA-4 | Melanoma | Ipilimumab (Yervoy®) | CHO | |
| Her2 | Breast cancer | Trastuzumab (Herceptin®) | CHO | |
| Pertuzumab (Perjeta®) | CHO | |||
| EGFR | Colorectal cancer | Cetuximab (Erbitux®) | SP2/0 | |
| Panitumumab (Vectibix®) | CHO | |||
| VEGF | Bevacizumab (Avastin®) | CHO | ||
| CD3/EpCAM | Malignant ascites | Catumaxomab# (Removab®) | CHO | |
| Anti-viral | A-epitope of viral fusion protein | Respiratory-Syncytial-Virus | Palivizumab (Synagis®) | CHO |
| B. anthracis protective antigen | Anthrax | Raxibacumab* (ABthrax®) | NS0 | |
| Others | C5 complement | Paroxysomal nocturnal hemoglobinuria | Eculizumab (Soliris®) | NS0 |
Not approved by *EMA/#FDA. mAbs withdrawn from the market are not included
Most therapeutic antibodies are chimeric (suffix –ximab; 70% human), humanized (suffix –zumab: 85-90% human) or human antibodies (suffix –umab: 100% human). These are less immunogenic compared with the initially used murine antibodies (suffix –omab: 100% mouse) [18]. IgGs are Y-shaped molecules and have a molecular weight of approx. 150kDa. They are composed of two “heavy” (approx. 50 kDa) and two “light” (approx. 25 kDa) polypeptide chains interconnected by disulfide bonds. The CH2 constant domain located on each heavy chain in the Fc region (= dimeric base of the antibody) has a conserved N-glycosylation consensus sequence at Asn297 (Figure 2). N-glycosylation is mandatory for Fc-effector functions of IgG which are mainly of anti-inflammatory nature such as the modulation of T- and NK (natural killer cell) activity [19]. One of the key immunogenic mechanisms of antibodies and a key element of antibodies used in cancer therapy is their ability for Fc-receptor binding. This antibody-receptor interaction can mediate compliment-dependent cytotoxicity (CDC) and antibody-dependent cellular cytotoxicity (ADCC) and can finally result in the lysis of the target cell. Αlpha-1,6-core-fucosylation of Fc N-glycans is a characteristic glycosylation feature of CHO cells and largely decreases the ability of the antibody to mediate ADCC [20]. Fucosyltransferase FUT 8 is responsible for the transfer of α1,6-linked fucose to the chitobiose core. The production of non-core-fucosylated anti-CD20 antibodies in CHO cells by FUT8 gene knockout resulted in a 100 fold increased ADCC [21].
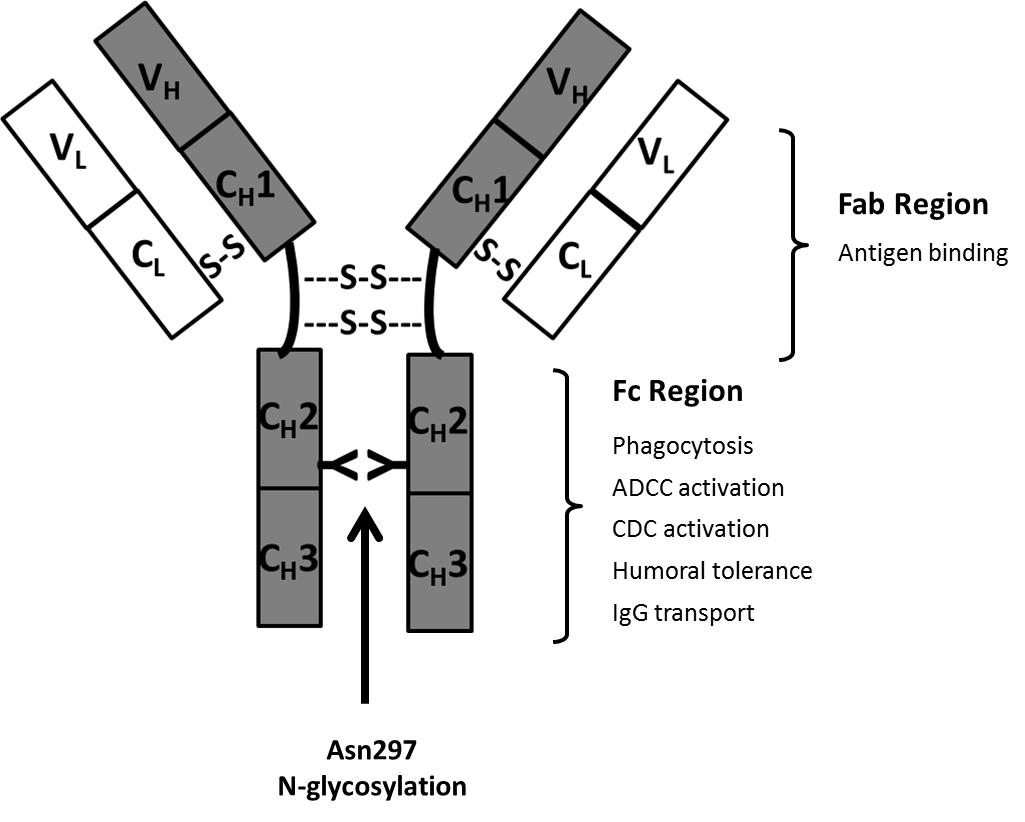
The antigen-binding sites of the antibody are located in its Fab region (i.e. arms of the antibody) which is composed of one constant and one variable domain from each heavy and light chain, respectively. Antibodies against a variety of target-antigens have been approved for therapeutic use. Most therapeutic antibodies lack Fab glycosylation. In a recent study six therapeutic mAbs (cetuximab, infliximab, basiliximab, palivizumab, panitumumab and zalutumumab) were investigated for glycosylation in the Fab region. However, glycosylation could only be confirmed for cetuximab [22]. Interestingly, hypersensitivity against cetuximab has been connected to the presence of the immunogenic α(1,3)-linked galactose residues in the Fab region of the antibody. This was mainly explained by the exposed nature of the Fab N-glycans as binding of IgE to α(1,3)-linked galactose in the Fc region of rituximab was absent [22]. Likewise, the increased complexity of Fab-glycosylation such as sialylation, galactosylation and the presence of Lewis-terminal structures can be explained by the exposure of this glycoprotein region during Golgi processing [23]. In therapeutic approaches which do not depend on Fc effector functions, antibody Fab fragments have been recognized as an attractive alternative [24]. Due to the lack of glycosylation these can be more cost-efficiently produced using bacterial expression systems. However, the absence of glycosylation results in a short serum half-life. Conversely, conjugation with polyethylene glycol (PEG) is a common strategy to prolong serum half-life [25]. The addition of 40kDa of PEG to a Fab fragment conferred a serum half-life similar to IgG [26]. Certolizumab pegol (Cimzia®) is an example of a PEGylated therapeutic mAb-Fab to tumor necrosis factor alpha (TNF-α) utilized in the treatment of Crohn's disease and rheumatoid arthritis.
Fc IgG fusion proteins
Therapeutic fusion proteins are created through joining genes from different proteins by recombinant DNA technology. This results in polypeptides which combine the properties of the originator proteins and often contain an additional linker peptide.
Examples of therapeutic Fc IgG fusion proteins include Alefacept (Amevive®), Abtacept (Orenci®), Belatacept (Nulojix®), Etanercept (Enbrel®) and Rilonacept (Arcalyst®) [27]. The most commercially successful fusion protein is the TNFα inhibitor Etanercept with global sales reaching $7.3 billion (USD) in 2010 [27]. Etanercept is a dimeric glycoprotein with a mass of approx. 150kDa and is used in the treatment of autoimmune diseases such as rheumatoid arthritis. It is composed of a human TNFα receptor part linked to an IgG1 Fc portion through an O-glycopeptide. Each part of the dimeric molecule carries one N-glycosylation site on its Fc part and two N-glycosylation sites on its TNFα unit. Figure 3 shows the total N-glycosylation profile of Etanercept (Enbrel®) as analyzed in our laboratory [5]. It includes a complex mixture of mono- to tetra-antennary core- and non-core-fucosylated structures which can carry up to two sialic acid residues. The glycan structures were identified by exo-enzymatic sequencing and confirmed by mass spectrometry as described in a later paragraph on glycan characterization [5]. By studying the N-glycosylation site heterogeneity of Etanercept (Enbrel®) we observed that the small biantennary neutral N-glycans were predominantly localized on the Fc part whereas larger tri- and tetra-antennary structures are attached to the TNFα unit [5]. Additionally, 12 occupied O-glycosylation sites carrying neutral, mono- and di-sialylated core 1 type structures were localized in the linker region of the fusion protein [5].
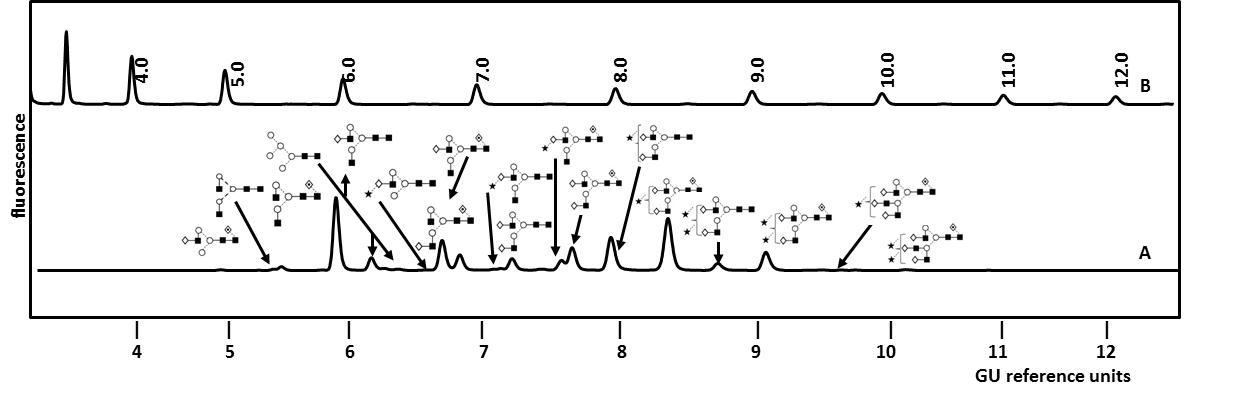
Figure 3.UPLC analysis of total N-linked glycans from Etanercept (Enbrel®) by HILIC-FLR as performed in our laboratory (A). The glycans were released from the fusion protein by PNGase F and fluorescently labelled. For an instrument-independent comparison, the retention times of peaks are transformed to standardized glucose unit values (GU) by comparing the profile to a dextran hydrolysate ladder (B). Structures were identified by sequential enzymatic digestion as exemplified in Figure 4 for the monosialylated N-glycan fraction from Etanercept (Enbrel®). Adapted from Houel et al [5], with permission from the American Chemical Society.
Erythropoietin
Erythropoietin (EPO) is a glycopeptide cytokine which controls and stimulates the production of red blood cells (termed erythropoiesis). The therapeutic use of EPO focusses on the restoration of blood haemoglobin concentration upon renal failure as well as the prevention of anemia in cancer patients undergoing treatement. Recombinantly produced therapeutic EPO is a glycopeptide with a mass of approx. 30kDa. It contains three N-linked glycosylation sites (Asn 24, 38, 83) which carry sialylated tetra-antennary structures and one O-glycosylation site (Ser 126) which carries mono- and disalylated core-1 structures [6]. The impact of glycosylation on EPO secretion, stability, half-life and effector functions has been extensively studied [28,29]. Full N-deglycosylation resulted in a total loss of EPO biological activity and a loss in resistance to thermal stress [28]. Sialylation plays an important role in serum half-life. De-sialylated EPO has a half-life of only 2 min and is subsequently rapidly cleared in the liver via galactosyl receptors of the hepatocytes [30]. Pharmaceutical EPO preparations form a large product family and are sub-divided into different classes based on their glycosylation characteristics. Epoetin-α (i.e. Epogen® and Eprex®) and epoetin-β (i.e. Recormon® and NeoRecormon®) are both produced in CHO cell systems but differ in their glycosylation characteristics [31]. It was shown that Eprex® (epoetin-α) has a higher degree of O-acetylation and a higher relative amount of immunogenic Neu5Gc per total sialic acid than NeoRecommon® (epoetin-β) [32, 33]. Epoetin-δ (Dynepo®, withdrawn from the market in 2008) is most similar to human EPO due to its production in human cell lines (HT-1080). Dynepo® has neither any Neu5Gc nor O-acetylation but is the only isoform which contains sialyl-Lewis x epitopes (SLex; [Fuc(α1-3)[Neu5Ac(α 2-3)Gal(β1-4)]GlcNAc(β-)]) [32].
Darbepoetin (Aranesp®) is a hypergalactosylated EPO-analogue for which two additional glycosylation sites (Asn 30, 88) have been introduced by glyco-engineering [34]. A comparison of darbepoetin alpha to convential epoetin-α/-β is given in Table 2. Darbepoetin can carry up to 22 sialic acid residues compared to just 14 in conventionally produced EPO. This results in an up to four times increased serum half-life and 2.2-fold higher in vivo activity [35-37]. However, the in vivo activity does not correlate with the receptor binding potential. The receptor binding correlates inversely with glycosylation and therefore requires the application of a six to 14-fold higher concentration of darbepoetin to achieve similar half-maximal receptor binding activity as EPO but allows longer dosing intervals due to its lower clearance rate [36,37].
Table 2. Comparison of the structural and pharmacokinetic properties of epoetin-α/-β and hypergalactosylated darbepoetin-α [31,32,35,36]
| Epoetin-α/-β | Darbepoetin-α | |
| Number amino acids | 165 | 165 |
| Glycosylation sites | 3x N-glycans (Asn 24, 38, 83)1x O-glycan (Ser 83) | 5x N-glycans (Asn 24, 30, 38, 83, 88)1x O-glycan (Ser 83) |
| Number sialic acid residues per molecule | up to 14 | up to 22 |
| Sialic acid O-acetylation | + | +++ |
| Glycan content per molecule | up to 40% | up to 51% |
| Molecular weight | 30.4 kDa | 37.1 kDa |
| Half-life (intravenous administration) | 6-9 h | 25h |
| Half-life (subcutaneous administration) | 19-24h | 48h |
| In vivo activity | 2.2 fold higher than epoetin-α/-β | |
| Dose requirement for half-maximalreceptor binding activity | 6-14x higher than epoetin-α/-β |
+++ major abundance + minor abundance
Other cytokines, growth factors, hormones, clotting factors and enzymes
Therapeutic interferons (IFNs) are glycoproteins of the cytokine family. Approved glycosylated IFNs include IFN-α (Alferon®/Avonex®) and IFN-β (Rebif®). IFN-α contains one potential glycosylation site. Upon introduction of an O-glycosylation site reduced thermal stability was observed whereas the introduction of four N-glycosylation sites resulted in improved serum half-life [38,39]. Likewise, a higher stability and in vitro availability were observed for glycosylated IFN-β (one N-glycosylation site) compared to non-glycosylated IFN-β [40].
Similar to IFNs, the haematopoietic growth factor granulocyte colony-stimulating factor (G-CSF) is available in its glycosylated (Lenograstim®) and non-glycosylated (Filgrastim®) form. G-CSF is a peptide hormone used in cancer therapy to reduce the risk of neutropenia. Glycosylated G-CSF is produced in CHO cells and carries one O-glycosylation site whereas non-glycosylated G-CSF is produced in E coli. Although in vitro studies depicted an up to 20-fold increase in activity of the glycosylated analog no differences were observed in vivo [41,42].
Therapeutic hormones, clotting factors and enzymes form large classes of therapeutic glycoproteins. The effects of glycosylation on the stability, in vivo efficacy and serum half-life of these glycoprotein classes were carefully reviewed by Sola et al. [43]. In additional studies, the introduction of four additional N-glycosylation sites on follitropin (Follistim® / Gonal F®), increased both the in vivo bioactivity and the serum half-life of the follicle-stimulating hormone up to twofold [44,45]. The introduction of N-glycosylation sites resulted in a higher biopotency compared to the introduction of O-glycosylation sites [45]. On the contrary, the affinity of the antithrombotic serine protease drotrecogin-α (Xigris®) to thrombin increased upon selective removal of one of the four N-glycosylation sites (Asn 313) [46]. Enzyme replacement therapy is applied in rare lysosomal storage diseases such as Fabry disease in which agalsidase-α (Replagal®) and agalsidase-β (Fabrazyme®) are used successfully. In this case the exposure of mannose/mannose-6-phosphate at the terminals of the six N-glycosylation sites on the glycoproteins are of great importance for the mannose-6-phosphate receptor mediated cellular internalisation of the enzyme [47].
Significance of protein glycosylation
Glycosylation can modulate the immunogenicity, efficacy, solubility and pharmacokinetic behavior of biopharmaceuticals and was extensively reviewed by Hossler et al and Walsh et al [2,4]. Multiple relations were reported between N-glycosylation and the therapeutic efficacy and immunogenicity of therapeutic proteins, while much less is known about the influence of O-glycans mainly attributed to their in-homogenous chemical nature.
Total N- and O-glycosylation
Glycan macro- and micro-heterogeneity can influence the folding, biological activity, kinetics and stability of therapeutic proteins. As observed for EPO, the total removal of N-glycans resulted in a significant decrease in product secretion, catabolic half-life and in vivo biological activity whereas the removal of the O-linked glycan did not have any effect [29]. Conversely, the glycoengineering of additional N-glycosylation sites on EPO, IFN-α and follitropin resulted in increased biological activity as well as increased serum half-life [38,44,45,48]. The lack of O-glycosylation on recombinant human granulocyte macrophage colony stimulating factor (rhGM-CSF) resulted in antigenicity and highlighted the role of O-glycans in masking potentially antigenic sites on the protein backbone [49].
Sialylation
Terminal N-glycan sialylation is an important quality parameter which determines the serum half-life of a protein. A 200-fold or more decrease in serum half-life of completely de-sialylated EPO compared to the sialylated reference EPO was observed when injected intravenously in rats [50]. Sialylation masks structural determinants such as mannose which are otherwise prone to ligand interaction and thus clearance of the molecule. Sialic acids on the Fc portion of intravenous gamma globulins have been shown to play an important role in the anti-inflammatory properties of the molecules as was demonstrated in a mouse model for serum arthritis [51]. The induction of inhibitory FcɣRIIB by macrophages which consequently leads to the therapeutically desired FcɣRIII activation in autoimmune diseases such as rheumatoid arthritis showed to be mediated by sialic acids on human intravenous ɣ globuline [52].
Sialic acid O-acetylation
O-acetylation of sialic acids has been recognized as an important quality parameter for erythropoiesis stimulating agents such as Eprex® and NeoRecormon® [32]. Due to the increased hydrophobicity and decreased susceptibility to sialidases conferred through O-acetylation an extension in the serum half-life can be assumed [32,53].
Galactosylation
The proportion of a-galactosylated (G0), mono-galactosylated (G1) and di-galactosylated (G2) N-glycans is dependent on cell culture conditions. CHO cells generally result in low galactosylation rates [54]. The assessment of terminal galactosylation is required by regulatory authorities. Terminal N-glycan galactosylation is directly related to N-glycan sialylation. The possible impact on CDC activity through involvement in complement C1q binding was shown for rituximab but, overall, variations in Fc galactosylation are not considered to adversely influence product stability or safety [54,55].
Mannosylation and terminal GlcNAc
Recognition of high mannose type N-glycans by mannose receptors and mannose binding lectins as well as the induction of endocytosis of the reticulo-endothelial system by terminal Man and GlcNAc promotes an accelerated serum clearance of the respective glycoproteins [56-58]. On the other hand it has been shown in vitro that antibodies carrying high-mannose structures (Man5, Man8/9) potentially enhance ADCC, decrease CDC and increase the binding affinity to FcɣRIIIa [59].
Core-fucosylation and bi-secting GlcNAc
Antibody-dependent cell-mediated cytotoxiticy (ADCC) is triggered by communication between IgG-Fc and natural killer (NK) cells and is mediated through the receptor FcɣRIIIa expressed on NK. Core α(1,6)-fucosylation of Fc N-glycans negatively affects this effector function [20]. The presence of core α(1,6)-fucosylation is inversely linked to the presence of the glycosyl transferase GnT-III, which is responsible for the addition of bisecting GlcNAc. Cell-engineering approaches which aim to inactivate core-fucosylation and simultaneously introduce GnT-III succeeded in increasing ADCC by almost 100 fold and thus is an essential step in the manufacturing of ADCC-mediating therapeutic proteins utilized in cancer therapy [60,61].
Non-human glycan epitopes
Non-human sugars on therapeutic proteins are a result of the production cell-line used and can lead to an immunological response. N-glycolyl-neuraminic acid (Neu5Gc) and terminal α(1,3)-linked galactose are xenoreactive sugars from mammalian cell-lines [62]. Candidate cell lines from yeasts, insects and transgenic plants contain additional immunogenic sugars such as α(1,3) core-fucose and β(1,6) xylose [62]. The pre-clinical assessment of xenoreactive sugars is complicated by their non-immunogenicity in animals. This requires the development of alternative test models such as CMAH (cytidine monophosphate-N-acetylneuraminic acid hydroxylase-like protein) knockout mice [63] which eliminate the biosynthesis of Neu5Gc from all cells mimicking the normal human lack of functional CMAH.
Neu5Gc is considered as an oncofetal antigen [64] and anti-Neu5Gc antibodies in humans have been shown to induce complement-mediated cytotoxiticy in the presence of Neu5Gc [65]. In the case of EPO, low levels of Neu5Gc (i.e. 1%), induced a negligible immunogenic response whereas levels of 7% of Neu5Gc showed a considerable response [66]. Additionally, up to 1% of total human circulating antibodies are directed against α(1,3)-linked galactose [67]. Cetuximab-induced anaphylaxis in some areas of the United States could be related to IgE specific for α(1,3)-linked galactose in patient sera [68].
Factors influencing protein glycosylation in biologics production
The glycosylation characteristics of therapeutic proteins are largely determined by culture systems and conditions. A detailed understanding of the production process and the monitoring of glycosylation during manufacturing is therefore required in order to assure product safety and efficiency.
Cell culture systems
Cellular expression systems which are capable of producing human-like N-glycosylation are essential for the manufacture of biopharmaceutical glycoproteins. Since bacterial expression systems (such as E.coli) lack the necessary enzymatic glycosylation machineries, the use of mammalian cell lines is common practice in glycoprotein production. Most frequently used are Chinese hamster ovary cells (CHO) followed by baby hamster kidney cells (BHK), murine myeloma (NS0) and hybridoma (SP2/0) cells, human cell lines, transgenic animals and milks (e.g. pig, goat). As mammalian cell expression systems are complex, often difficult to scale up and expensive, alternative production platforms based on plants, yeasts, and insects are currently under investigation. An overview on the glycosylation characteristics of cellular expression systems is given in Table 3 and has been extensively reviewed and studied [16,69-73]. The main disadvantages of CHO, NS0, SP2/0 and transgenic animals/milks is the presence of non-human Neu5Gc and α(1,3) Gal epitope [62,74] which can evoke immunogenic responses in humans. Another critical point is the presence of core-(α1,6) fucosylation which reduces the ADCC effector function of mAbs. CHO cells lack GNT-III which is responsible for the addition of bi-secting GlcNAc and results in the increased presence of core-(α1,6) fucosylation instead. Glyco-engineering approaches aimed at decreasing the glycoprotein immunogenicity and the overexpression of GNT-III are ongoing [75,76]. The use of human cell lines (e.g. Per.C6, Hek293, HT-1080) is of special interest because their glycosylation machinery closest resembles that of humans. Additionally, a tenfold increased productivity was observed in the human cell line Per.C6 compared to conventional cell lines [77]. However, their application for commercial manufacturing is still limited as the risk of pathogenic infection presents an additional hurdle for regulatory approval.
Table 3. Glycosylation characteristics of standard and alternative production cell lines. According to [16,70-73] and research performed in our laboratory
| CHO | BHK | NS0, SP2/0 | human | animal&milk | plant | yeast | insect | bacterial | |
| glycosylation | + | + | + | + | + | + | + | + | - |
| sialylation | ++ | + | +++ | ++ | ++ | - | - | - | - |
| α2,6-sialyl | - | - | + | + | +/- | - | - | - | - |
| Neu5Gc | ++ | +++ | +++ | +/-* | +++ | - | - | - | - |
| α1,3-Gal | + | + | ++ | - | ++ | - | - | - | - |
| bisect. GlcNAc | - | - | - | + | +/- | - | - | - | - |
| α1,6-core Fuc | + | + | ++ | + | + | +/- | - | + | - |
| α1,3-core Fuc | - | - | - | - | - | + | - | + | - |
| β1,6-xylose | - | - | - | - | - | + | - | - | - |
| high-mannose | + | + | + | + | + | + | +++ | ++ | - |
| pauci-mannose | - | - | - | - | - | - | - | +++ | - |
| additional characteristics | - | - | - | outer-armfucosylation (Lex/SLex) | - | outer-armfucosylation(Lea) | Phosphorylation | - | - |
| approvedtherapeutics(examples) | Enbrel®(Etanercept) | Helixate®(FS Factor VIII) | Arzerra ® (Ofatumumab)a | Elaprase® (idursulfase) | ATryn® (antithrombin) | - | - | - | Neupogen® (filgrastim) |
| Rituxan®(Rituximab) | NovoSeven® (Factor VIIa) | Remicade® (Infliximab)b | Xigris® (Drotrecoginα) | Creon® (pancrelipase) | - | - | - | Humalog® (insulin) | |
+++ abundant presence ++ presence + low presence – not present +/- both, presence and absence reported. *presence of Neu5Gc from exogenous sources possible; aNS0; bSP2/0. Lex: Lewis x epitope [Fuc(α1-3)[Gal(β1-4)]GlcNAc(β-)]. SLex: sialyl-Lewis x epitope [Fuc(α1-3)[Neu5Ac(α 2-3)Gal(β1-4)]GlcNAc(β-)]. Lea: Lewis a epitope [Gal(β1-3) [Fuc(α1-4)]GlcNAc(β-)]
No therapeutic proteins produced by plants, yeasts or insects have been approved thus far despite their high productivity, low costs and ease of scaling up to industrial production levels [62]. Reasons for this include the presence of immunogenic sugars such as α(1,3) core-fucosylation and β(1,6) xylose. Additionally, the overall lack of sialylation and the presence of highly mannosylated structures ranging up to Man100GlcNAc2 for yeasts and paucimannosic structures (i.e. Man3GlcNAc2) lead to an accelerated clearance of these glycoproteins [62]. Significant efforts have been made in glycoengineering of alternative production cell lines resulting for example in the successful production of complex N-glycans in Drosophila S2 cells or the purification of rMAbs carrying human N-glycosylation from yeast cells [78,79].
Cell culture conditions
During the development and production of biopharmaceuticals a continuous scaling- up of the manufacturing process is required. Scaling up the production of biopharmaceuticals is a complex task as the increase of protein quantity might affect and compromise product quality [80]. Changes in process therefore require thorough comparability studies which need to be performed according to comparability protocols defined by the regulatory authorities [81]. Due to concerns in concomitant changes in glycan structures the FDA recently banned the application of up-scaling in the production of aglucosidase α (Myozyme®) from 160 L scale to 2000 L scale and required a new biologic license for the application [82].
The manufacturing mode and processing variables such as pH, temperature, oxygen level and media composition have a significant effect on protein glycosylation which was extensively reviewed by Hossler et al and is briefly summarized in the following paragraph [4]. Production methods applying low shear force (i.e. perfusion mode) generally result in slower cell growth but more complete glycosylation and overall sialylation [83,84]. A pH range of 6.8-7.2 was considered optimal for appropriate galactosylation and sialylation of EPO production in CHO cells [85,86]. Increased ammonia levels result in an increased culture pH and has been shown to have adverse effects on protein sialylation [87]. Similarly, a decrease in process temperature (i.e. from 37°C to 30°C) has been shown to negatively affect EPO sialylation although it resulted in an increased cell viability [85]. However, a synergistic effect between temperature and pH was observed since the loss of sialylation efficiency during lowered temperatures could be prevented by a simultaneous decrease in pH [85]. For a consistent glycosylation level, the degree of dissolved oxygen should be kept between 10 and 100% [88]. Critical media components include monosaccharides, nucleotide sugar precursors, small molecules such as sodium butyrate, ammonia and amino acids as well as lipids and metal ions. Cultures limited in monosaccharide supply show lowered glycosylation and sialylation whereas product- and culture-dependent trends were observed with nucleotide sugar supplementation [89-91]. Appropriate supplementation of amino acids, manganese and lipids were shown to have a positive impact on sialylation and N-glycan site occupancy for both recombinant human EPO and IFN-ɣ [92,93].
Furthermore, media composition can greatly influence the concentration of immunogenic sugars. For example, it was demonstrated that with the addition of sodium butyrate in CHO cell lines a reduction of Neu5Gc of 50-60% can be obtained [94].
Glycan characterization
The complexity of protein glycosylation means their analytical characterization is hugely challenging and usually requires the use of orthogonal techniques. N-glycans are most commonly analyzed after the enzymatic release by peptide N-glycosidase F (PNGaseF) using high- and ultra-performance liquid chromatography (HPLC/UPLC) or capillary electrophoresis (CE) coupled to a fluorescent detector and/or mass spectrometer (MS) [5,95,96]. Hydrophilic interaction liquid chromatography (HILIC) and reversed phase (RP) are stationary phases which can be used for complimentary chromatographic glycan separation whereas weak anion exchange (WAX) chromatography allows the separation of glycans according to their charge [97]. Fluorescent detection enables structural quantification but requires glycan derivatization by fluorescent labels (2-aminobenzamide [2-AB] and 2-aminobenzoic acid [2-AA] for HPLC/UPLC and 1-aminopyrene-3,6,8-trisulfonic acid [APTS] for CE-LIF) [97]. Matrix-assisted laser desorption-ionization (MALDI-TOF MS) and electrospray ionization (ESI) are routinely used for compositional glycan analysis [98]. Combined with mass fragmentation (MS/MS) these methods result in glycan sequence information based on the formation of diagnostic ions. Another powerful tool for glycan sequence and linkage identification which is routinely used in our laboratory is the enzymatic panel digestion of 2-AB labelled glycans using an array of linkage-specific sialidases, fucosidases, galactosidases, N-acetylhexosaminidases and mannosidase [97]. Figure 4 exemplifies our analytical workflow for the exo-enzymatic sequencing of mono-sialylated N-glycans obtained by WAX fractionation of Etanercept (Enbrel®) (see Figure 3 for the total N-glycan profile) which allowed the confident assignment of the structures present [5].
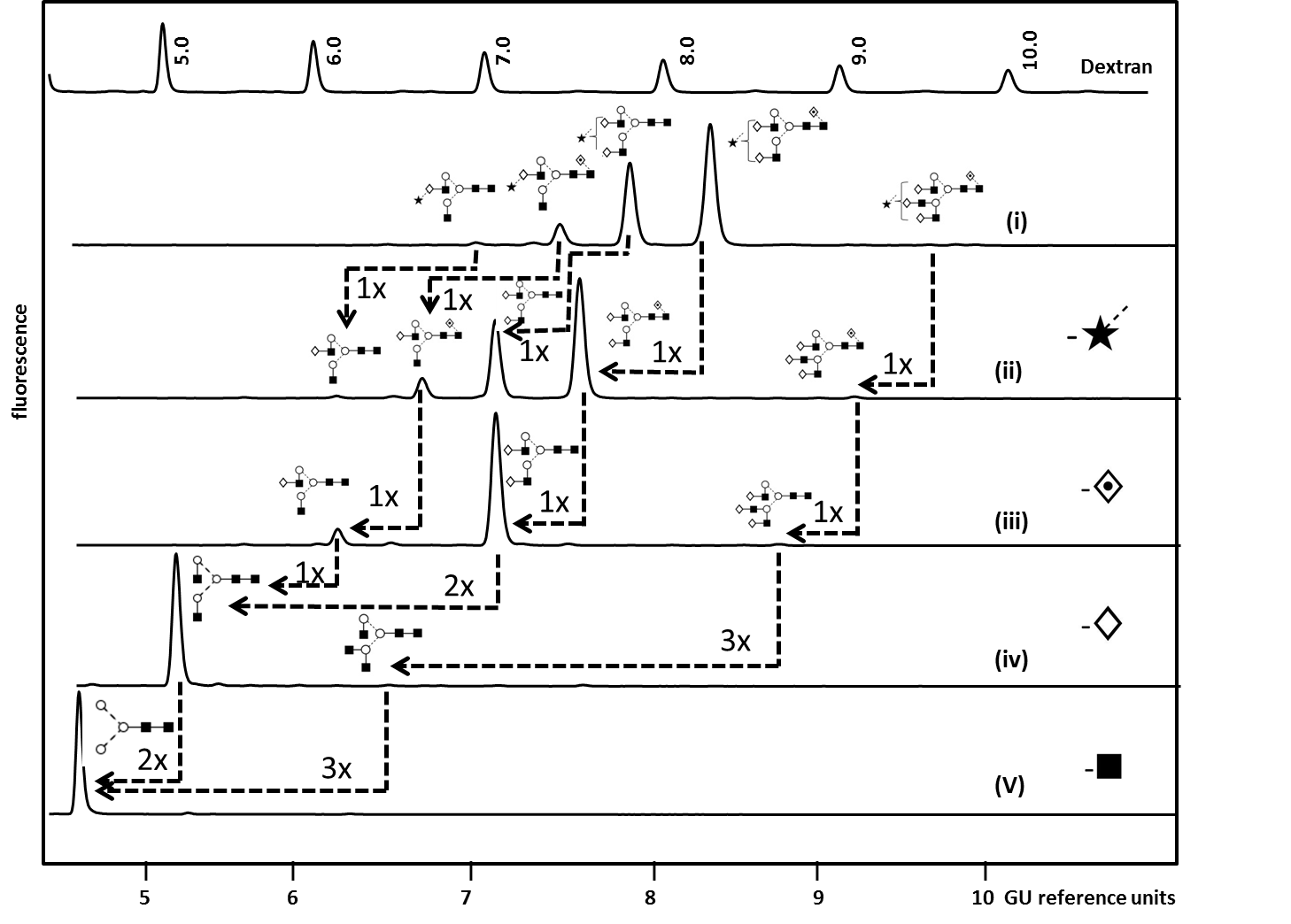
Figure 4.Exoglycosidase array analysis of fluorescently labelled mono-sialylated WAX fraction from Etanercept (Enbel®) by HILIC-fluorescence to determine N glycans structures. (i) Whole N-glycan pool released by PNGase F (ii) ABS (Athrobacter ureafaciens Sialidase) releases α2-3/6/8 sialic acids. (iii) BFK (Fucosidase from bovine kidney) releases α1-2/6 fucose. (iv) BTG (Bovine testes ß-galactosidase) releases galactose β1-3/4 linkages and (v) GUH (hexosaminidase) release β-GlcNAc but not GlcNAc linked to β1-4 Man.Dashed arrows indicate the enzymatic removal of one (1x), two (2x) or three (3x) sugar units. For an instrument-independent comparison, the retention times of peaks are transformed to standardized glucose unit values (GU) by comparing the profile to a dextran hydrolysate ladder. Adapted from Houel et al [5], with permission from the American Chemical Society
Proteolytic cleavage of IgG by using the enzyme IdeS FabRICATIOR facilitates the identification of glycan heterogeneity in the Fab and Fc region of monoclonal antibodies [5]. GlycoBase and UniCarbDB are two powerful structural databases which have been implemented for the efficient interpretation of LC and MS data [99, 100]. Furthermore, a low-cost robotic sample preparation platform has recently been established for the high-throughput IgG N-glycan analysis which results in highly reproducible data and considerably reduces manual sample handling errors [101]. The identification of O-glycans is more challenging due to the lack of a single consensus sequence for glycan attachment and the lack of a common core structure. The combination of Collision Induced Dissociation (CID), which results in glycan sequence information and Electron Transfer Dissociation (ETD), which enables the identification of the amino acid residue at the glycosylation site presents a powerful MS glycan characterization approach for simple O-glycans as present on therapeutic glycoproteins [5]. Similarly, peptide mapping is used to confirm glycosylation site occupancy in N-glycan analysis while reduced CE-SDS is used as a tool for assessing total glycosylation of protein sub-units [102].
Alterations in PTMs, and thus glycosylation, can affect the higher-order structure of proteins and was reviewed by Berkowitz et al [103]. This can, for example, result in protein aggregation. Global information on protein structure can be obtained by classical analytical techniques such as circular dichorism (CD), differential scanning calorimetry, analytical ultracentrifugation (AUC) and size exclusion chromatography [103]. Hydrogen-deuterium exchange (HDX) is a new and powerful MS based method which even enables the localization of these structural changes in biopharmaceuticals [104].
Assessment of protein glycosylation in biologics and biosimilar manufacturing
The characterization of protein glycosylation plays an important role during the production process of biologics and biosimilars. In biosimilar production the demonstration of similarity to the innovator product is a key regular requirement. Similarly, the manufacturer of biologics has to prove a reproducible and consistent production process and ensure that the desired glycosylation is present and immunogenic epitopes are reduced to a minimum. An example of the importance of controlled production are the “epidemic” incidences of Eprex®-induced antibody-induced red blood cell aplasia (PRCA) which was, amongst other factors, deduced to production-related changes in the carbohydrate profile [105,106]. Glycosylation changes between different producers, different production charges and biosimilar versus originator products were repeatedly observed [102,107-110].Differences were observed between the abundance of non-core-fucosylated N-glycans of rituximab and proposed biosimilar Rituximab GP2013 [102]. However, complementary CDC-, ADCC- and receptor binding assays of innovator and biosimilar product showed very comparable results. Differences in N-glycan galactosylation levels were observed between trastuzumab and a candidate biosimilar and changes in N-glycosylation site occupancy were observed between tenecteplase tissue plasminogen activator (TNK-tPA) and a follow-on product [107,110]. Despite the probable effect of the significantly decreased site-occupancy on the bioactivity of the biosimilar the follow-on TNK-tPa was considered acceptable for marketing [107,110].
Interesting observations were made by Kawasaki et al when comparing three epoetin α and one epoetin β products from two different countries [108]. Although for all products tetra-sialylated tetra-antennary structure were most abundant, significant intra-class differences were observed in the acetylation pattern and presence of smaller structures for epoetin α.
The pre- and post-production change variability of glycosylation attributes was recently studied for darbepoetin-α (Aranesp®), rituximab (Rituxan®/Mabthera®) and etanercept (Enbrel®) [109]. Significant decreases in darbepoetin-α sialylation by 10%, a 3-fold increase in non-core-fucosylated G0 for rituximab or a 20% decrease in the di-galactosylated structure G2F for etanercept did not result in a market withdrawal of the products.
The question as to which changes in glycosylation attributes are acceptable can thus only be answered on a case-by-case basis and should be done in combination with complimentary data.
Conclusion
The inherent variability of biological systems challenges the manufacturing process of biologics and biosimilars. The requirements of manufacturing biologics are complex and a thorough understanding of the product is crucial. It is therefore of importance that biomanufacturing follows quality-by-design (QbD) principles. QbD defines the critical quality attributes (CQAs) of a product and requires the understanding of the association between CQAs and clinical properties [111]. Glycosylation is a key quality attribute as it can influence production rate, efficacy and safety of pharmaceutical proteins. The approval of biosimilars and the approval of changes in the manufacturing process of biologics are strictly regulated. However, the decision of comparability is difficult and cannot be generalized. A state-of-the art analytical toolbox is a key requirement for the establishment of a more targeted development process. However, the availability and extent of reference-product batches might be limited to the manufacturer. The assessment of comparability and the refining of regulatory guidelines would therefore be greatly facilitated by the existence of data collections on commercialized biologics. The establishment of defined reference standards which integrate knowledge on structural characteristics and structure-function relationships is still a matter of debate [112] but will definitely move forward the quality of biologics and biosimilar legislation and production.
Acknowledgements
The work in our laboratory is supported by the European Commission under the Seventh Framework Programme (FP7) GlycoHIT [grant number 260600], GlycoBioM [grant number 259869], HighGlycan [grant number 278535], GastricGlycoExplorer [grant number 316929], by the Science Foundation Ireland (Alimentary Glycoscience Research Cluster (AGRC) [grant number 08/SRC/B1393]). We thank the NIBRT GlycoSciences group for the scientific discussion related to this paper and Dr Ciara McManus (NIBRT) for critically reading the manuscript.
Abbreviations
ADCC, antibody-dependent cellular cytotoxicity; Asn, asparagine; BHK, baby hamster kidney; BLys, B-lymphocyte stimulator; CD, cluster of differentiation; CDC, complement dependent cytotoxicity; CHO, Chinese hamster ovary; CTLA, cytotoxic T-lymphocyte-associated antigen; EGFR, epidermal growth factor receptor; EMA, European medicines agency; EpCAM, epithelial cell adhesion molecule; EPO, erythropoietin; FDA, food and drug administration; Fuc, fucose; Gal, galactose; G-CSF, growth factor granulocyte colony-stimulating factor;Glc, glucose; GlcNAc, N-acetyl-glucosamine; Her, human epidermal growth factor; HILIC-FLR, hydrophilic interaction liquid chromatography with fluorescence detection; IL, interleukin; IgG/E, immunoglobulin G/E; mAb, monoclonal antibody; Man, mannose; Neu5Ac, N-acetyl-neuraminic acid; Neu5Gc, N-glycolyl-neuraminic acid; PNGaseF, peptide N-glycosidase F; RANKL, receptor activator of nuclear factor-kappa B; Ser, serine; Thr, threonine; (S)Lea/x, (sialyl) Lewis a/x epitope; TNF, tumor necrosis factor; UPLC, ultra-performance liquid chromatography; VEGF, vascular endothelial growth factor; WAX, weak anion exchange chromatography.
- EvaluatePharma: World preview 2013, outlook to 2018: Returning to growth
- Walsh G., Jefferis R. Post-translational modifications in the context of therapeutic proteins. Nat Biotech 2006; 24: 1241-1252
- Dennis JW., Granovsky M., Warren CE. Protein glycosylation in development and disease. BioEssays 1999; 21: 412-421
- Hossler P., Khattak SF., Li ZJ. Optimal and consistent protein glycosylation in mammalian cell culture. Glycobiology 2009; 19: 936-949
- Houel S., Hilliard M., Yu YQ., McLoughlin N., Martin SM., Rudd PM., Williams JP., Chen W. N- and O-glycosylation analysis of etanercept using liquid chromatography and quadrupole time-of-flight mass spectrometry equipped with electron-transfer dissociation functionality. Anal Chem 2013; 86: 576-584
- Jensen PH., Karlsson NG., Kolarich D., Packer NH. Structural analysis of N- and O-glycans released from glycoproteins. Nat Protocols 2012; 7: 1299-1310
- Tsang L., Cortez N., Gad SC. Biopharmaceuticals: Definition and regulation. in: Pharmaceutical sciences encyclopedia, John Wiley & Sons, Inc., New York, 2010; 1-18
- Williams PD. Methods of production of biopharmaceutical products and assessment of environmental impact. in: Preclinical safety evaluation of biopharmaceuticals, John Wiley & Sons, Inc., New York, 2007; 21-41
- Beck A., Reichert JM. Approval of the first biosimilar antibodies in Europe: A major landmark for the biopharmaceutical industry. MAbs 2013; 5: 621-623
- Dalgaard K., Evers M., da Silva JS. Biosimilars seven years on: Where are we and what's next? McKinsey&Company 2013: 1-9
- Shaughnessy AF. Monoclonal antibodies: Magic bullets with a hefty price tag. BMJ 2012; 345: e8346
- De la Horie, GFC. Making biologic drugs more affordable. 2010. Available from: www.dddmag.com/articles/2010/07/making-biologic-drugs-more-affordable; [Accessed: 31.01.2014]
- GaBI online - generics and biosimilars initiative.Biosimilars used in Europe. 2014. Available from: www.gabionline.net/Biosimilars/General/Biosimilars-approved-in-Europe; [Accessed: 04.02.2014]
- European medicines agency, committee for medicinal products for human use (CHMP). Guideline on similar biological medicinal products containing monoclonal antibodies - non-clinical and clinical issues 2012 [cited data: 31.01.2014]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500128686.pdf; [Accessed: 31.01.2014]
- Food and drug administration, center for drug evaluation and research (CDER). Guidance for industry.Scientific considerations in demonstrating biosimilarity to a reference product. 2012. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291128.pdf; [Accessed: 31.01.2014]
- Ghaderi D., Zhang M., Hurtado-Ziola N., Varki A. Production platforms for biotherapeutic glycoproteins. Occurrence, impact, and challenges of non-human sialylation. Biotechnol Genet Eng Rev 2012; 28: 147-175
- Leavy O. Therapeutic antibodies: Past, present and future. Nat Rev Immunol 2010; 10: 297
- Hudson P.J., Souriau C.: Engineered antibodies. Nat Med 2003; 9: 129-134
- Abès R., Teillaud J.-L. Impact of glycosylation on effector functions of therapeutic IgG. Pharmaceuticals 2010; 3: 146-157
- Okazaki A., Shoji-Hosaka E., Nakamura K., Wakitani M., Uchida K., Kakita S., Tsumoto K., Kumagai I., Shitara K. Fucose depletion from human IgG1 oligosaccharide enhances binding enthalpy and association rate between IgG1 and FcγRIIIa. J Mol Biol 2004; 336: 1239-1249
- Yamane-Ohnuki N., Kinoshita S., Inoue-Urakubo M., Kusunoki M., Iida S., Nakano R., Wakitani M., Niwa R., Sakurada M., Uchida K., Shitara K., Satoh M. Establishment of FUT8 knockout chinese hamster ovary cells: An ideal host cell line for producing completely defucosylated antibodies with enhanced antibody-dependent cellular cytotoxicity. Biotechnol Bioeng 2004; 87: 614-622
- van Bueren JJL., Rispens T., Verploegen S., van der Palen-Merkus T., Stapel S., Workman LJ., James H., van Berkel PHC., van de Winkel JGJ., Platts-Mills TAE., Parren PWHI. Anti-galactose-[alpha]-1,3-galactose IgE from allergic patients does not bind [alpha]-galactosylated glycans on intact therapeutic antibody Fc domains. Nat Biotech 2011; 29: 574-576
- Jefferis R. Glycosylation of recombinant antibody therapeutics. Biotechnol Prog 2005; 21: 11-16
- Nelson AL. Antibody fragments: Hope and hype. MAbs 2010; 2: 77-83
- Jain A., Jain SK. PEGylation: An approach for drug delivery. A review. Crit Rev Ther Drug Carrier Syst 2008; 25: 403-47
- Chapman AP., Antoniw P., Spitali M., West S., Stephens S., King DJ. Therapeutic antibody fragments with prolonged in vivo half-lives. Nat Biotech 1999; 17: 780-783
- Reichert JM. Therapeutic Fc-fusion proteins and peptides as successful alternatives to antibodies. MAbs 2011; 3: 415-416
- Tsuda E., Kawanishi G., Ueda M., Masuda S., Sasaki R. The role of carbohydrate in recombinant human erythropoietin. Europ J Biochem 990; 188: 405-411
- Wasley L., Timony G., Murtha P., Stoudemire J., Dorner A., Caro J., Krieger M., Kaufman R. The importance of N- and O-linked oligosaccharides for the biosynthesis and in vitro and in vivo biologic activities of erythropoietin. Blood 1991; 77: 2624-2632
- Spivak JL., Hogans BB. The in vivo metabolism of recombinant human erythropoietin in the rat. Blood 1989; 73: 90-9
- Jelkmann W. Recombinant EPO production—points the nephrologist should know. Nephrol Dial Transplant 2007; 22: 2749-2753
- Shrokh Z., Royle L., Saldova R., Bones J., Abrahams JL., Artemenko NV., Flatman S., Davies M., Baycroft A., Sehgal S., Heartlein MW., Harvey DJ., Rudd PM. Erythropoietin produced in a human cell line (Dynepo) has significant differences in glycosylation compared with erythropoietins produced in CHO cell lines. Mol Pharm 2010; 8: 286-296
- Storring PL., Tiplady RJ., Gaines Das RE., Stenning BE., Lamikanra A., Rafferty B., Lee J. Epoetin alpha and beta in their erythropoetin isoform compositions and biological properties. Brit J Haematol 1998; 100: 79-89
- Egrie JC., Browne JK. Development and characterization of novel erythropoiesis stimulating protein (NESP). Nephrol Dial Transplant 2001; 16 Suppl 3: 3-13
- Osterborg A. New erythropoietic proteins: Rationale and clinical data. Semin Oncol 2004; 31: 12-8
- Jelkmann W. Recombinant erythropoietins – the role of glycosylation in receptor binding, action and degradation. Business Briefing: European Kidney & Urological Disease B 2006: 1-5
- Elliott S., Egrie J., Browne J., Lorenzini T., Busse L., Rogers N., Ponting I. Control of rHuEPO biological activity: The role of carbohydrate. Exp Hematol 2004; 32: 1146-1155
- Ceaglio N., Etcheverrigaray M., Kratje R., Oggero M. Influence of carbohydrates on the stability and structure of a hyperglycosylated human interferon alpha mutein. Biochimie 2010; 92: 971-978
- Johnston M., Frahm G., Li X., Durocher Y., Hefford M. O-linked glycosylation leads to decreased thermal stability of interferon alpha 2b as measured by two orthogonal techniques. Pharm Res 2011; 28: 1661-1667
- Runkel L., Meier W., Pepinsky RB., Karpusas M., Whitty A., Kimball K., Brickelmaier M., Muldowney C., Jones W., Goelz SE. Structural and functional differences between glycosylated and non-glycosylated forms of human interferon-beta (IFN-beta). Pharm Res 1998; 15: 641-649
- Bonig H., Silbermann S., Weller S., Kirschke R., Korholz D., Janssen G., Gobel U., Nurnberger W. Glycosylated vs non-glycosylated granulocyte colony-stimulating factor (G-CSF)--results of a prospective randomised monocentre study. Bone Marrow Transplant 2001; 28: 259-64
- Nissen C. Glycosylation of recombinant human granulocyte colony stimulating factor: Implications for stability and potency. Eur J Cancer 1994; 30A Suppl 3: S12-4
- Solá R., Griebenow K. Glycosylation of therapeutic proteins. BioDrugs 2010; 24: 9-21
- Perlman S., van den Hazel B., Christiansen J., Gram-Nielsen S., Jeppesen CB., Andersen KV., Halkier T., Okkels S., Schambye HT.: Glycosylation of an N-terminal extension prolongs the half-life and increases the in vivo activity of follicle stimulating hormone. J Clin Endocrinol Metab 2003; 88: 3227-3235
- Weenen C., Peña JE., Pollak SV., Klein J., Lobel L., Trousdale RK., Palmer S., Lustbader EG., Ogden RT., Lustbader JW. Long-acting follicle-stimulating hormone analogs containing N-linked glycosylation exhibited increased bioactivity compared with O-linked analogs in female rats. J Clin Endocrinol Metab 2004; 89: 5204-5212
- Grinnell BW., Walls JD., Gerlitz B. Glycosylation of human protein C affects its secretion, processing, functional activities, and activation by thrombin. J Biol Chem 1991; 266: 9778-9785
- Barbey F., Hayoz D., Widmer U., Burnier M. Efficacy of enzyme replacement therapy in Fabry disease. Curr Med Chem Cardiovasc Hematol Agents 2004; 2: 277-286
- Gegrie J.C., Browne J.K. Development and characterization of novel erythropoiesis stimulating protein (NESP). Br J Cancer 2001; 84: 3-10
- Gribben JG., Devereux S., Thomas NSB., Keim M., Jones HM., Goldstone AH., Linch DC. Development of antibodies to unprotected glycosylation sites on recombinant human GM-CSF. Lancet 1990; 335: 434-437
- Erbayraktar S., Grasso G., Sfacteria A., Xie Q.-w., Coleman T., Kreilgaard M., Torup L., Sager T., Erbayraktar Z., Gokmen N., Yilmaz O., Ghezzi P., Villa P., Fratelli M., Casagrande S., Leist M., Helboe L., Gerwein J., Christensen S., Geist M.A., Pedersen L.Ø., Cerami-Hand C., Wuerth J.-P., Cerami A., Brines M. Asialoerythropoietin is a nonerythropoietic cytokine with broad neuroprotective activity in vivo. P Natl Acad Sci 2003; 100: 6741-6746
- Kaneko Y., Nimmerjahn F., Ravetch J.V. Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science 2006; 313: 670-673
- Bruhns P., Samuelsson A., Pollard JW., Ravetch JV. Colony-stimulating factor-1-dependent macrophages are responsible for IVIG protection in antibody-induced autoimmune disease. Immunity 2003; 18: 573-58
- Argueso P., Sumiyoshi M. Characterization of a carbohydrate epitope defined by the monoclonal antibody H185: Sialic acid O-acetylation on epithelial cell-surface mucins. Glycobiology 2006; 16: 1219-1228
- Raju TS., Jordan RE. Galactosylation variations in marketed therapeutic antibodies. MAbs 2012; 4: 385-91
- Hodoniczky J., Zheng YZ., James DC. Control of recombinant monoclonal antibody effector functions by Fc N-glycan remodeling in vitro. Biotechnol Progr 2005; 21: 1644-1652
- Malhotra R., Wormald MR., Rudd PM., Fischer PB., Dwek RA., Sim RB. Glycosylation changes of IgG associated with rheumatoid arthritis can activate complement via the mannose-binding protein. Nat Med 1995; 1: 237-243
- Wileman TE., Lennartz MR., Stahl PD. Identification of the macrophage mannose receptor as a 175-kDa membrane protein. Proc Natl Acad Sci U S A 1986; 83: 2501-2505
- Maynard Y., Baenziger JU. Oligosaccharide specific endocytosis by isolated rat hepatic reticuloendothelial cells. J Biol Chem 1981; 256: 8063-8
- Yu M., Brown D., Reed C., Chung S., Lutman J., Stefanich E., Wong A., Stephan JP., Bayer R. Production, characterization, and pharmacokinetic properties of antibodies with N-linked mannose-5 glycans. MAbs 2012; 4: 475-487
- Shields RL., Lai J., Keck R., O'Connell LY., Hong K., Meng YG., Weikert SHA., Presta LG. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human FcγRIII and antibody-dependent cellular toxicity. J Biol Chem 2002; 277: 26733-26740
- Davies J., Jiang L., Pan L-Z., LaBarre MJ., Anderson D., Reff M. Expression of GnTIII in a recombinant anti-CD20 CHO production cell line: Expression of antibodies with altered glycoforms leads to an increase in ADCC through higher affinity for FCγRIII. Biotechnology and Bioengineering 2001; 74: 288-294
- Ghaderi D., Taylor RE., Padler-Karavani V., Diaz S., Varki A. Implications of the presence of N-glycolylneuraminic acid in recombinant therapeutic glycoproteins. Nat Biotech 2010; 28: 863-867
- Hedlund M., Tangvoranuntakul P., Takematsu H., Long JM., Housley GD., Kozutsumi Y., Suzuki A., Wynshaw-Boris A., Ryan AF., Gallo RL., Varki N., Varki A. N-glycolylneuraminic acid deficiency in mice: Implications for human biology and evolution. Mol Cell Biol 2007; 27: 4340-4346
- Varki A. N-glycolylneuraminic acid deficiency in humans. Biochimie 2001; 83: 615-622
- Nguyen DH., Tangvoranuntakul P., Varki A. Effects of natural human antibodies against a nonhuman sialic acid that metabolically incorporates into activated and malignant immune cells. J Immunol 2005; 175: 228-236
- Noguchi A., Mukuria CJ., Suzuki E., Naiki M. Immunogenicity of N-glycolylneuraminic acid-containing carbohydrate chains of recombinant human erythropoietin expressed in chinese hamster ovary cells. J Biochem 1995; 117: 59-62
- Galili U., Anaraki F., Thall A., Hill-Black C., Radic M. One percent of human circulating B lymphocytes are capable of producing the natural anti-Gal antibody. Blood 1993; 82: 2485-2493
- Chung CH., Mirakhur B., Chan E., Le Q.-T., Berlin J., Morse M., Murphy B.A., Satinover SM., Hosen J., Mauro D., Slebos RJ., Zhou Q., Gold D., Hatley T., Hicklin DJ., Platts-Mills TAE. Cetuximab-induced anaphylaxis and IgE specific for galactose-α-1,3-galactose. New England Journal of Medicine 2008; 358: 1109-1117
- Houdebine L.-M. Antibody manufacture in transgenic animals and comparisons with other systems. Curr Opin Biotech 2002; 13: 625-629
- Struwe WB., Cosgrave EFJ., Byrne JC., Saldova R., Rudd PM. Glycoproteomics in health and disease. in: Functional and structural proteomics of glycoproteins, Owens RJ., Nettleship JE., Springer, Dordrecht, 2011; 1-38
- Swiech K., Picanço-Castro V., Covas D.T. Human cells: New platform for recombinant therapeutic protein production. Protein Expr Purif 2012; 84: 147-153
- Rendic D., Wilson IBH., Paschinger K. The glycosylation capacity of insect cells. CCACAA 2008; 81: 7-21
- Wilson IBH., Zeleny R., Kolarich D., Staudacher E., Stroop CJM., Kamerling JP., Altmann F. Analysis of Asn-linked glycans from vegetable foodstuffs: Widespread occurrence of Lewis a, core α1,3-linked fucose and xylose substitutions. Glycobiology 2001; 11: 261-274
- Bosques CJ., Collins BE., Meador JW., Sarvaiya H., Murphy JL., DelloRusso G., Bulik DA., Hsu IH., Washburn N., Sipsey SF., Myette JR., Raman R., Shriver Z., Sasisekharan R., Venkataraman G. Chinese hamster ovary cells can produce galactose-[alpha]-1,3-galactose antigens on proteins. Nat Biotech 2010; 28: 1153-1156
- Sburlati AR., Umaña P., Prati E.G.P., Bailey JE. Synthesis of bisected glycoforms of recombinant IFN-β by overexpression of β-1,4-N-Acetylglucosaminyltransferase III in chinese hamster ovary cells. Biotechnol Progr 1998; 14: 189-192
- Ferrara C., Brünker P., Suter T., Moser S., Püntener U., Umaña P. Modulation of therapeutic antibody effector functions by glycosylation engineering: Influence of Golgi enzyme localization domain and co-expression of heterologous β1, 4-N-acetylglucosaminyltransferase III and Golgi α-mannosidase II. Biotech Bioeng 2006; 93: 851-861
- Coco-Martin JM., Harmsen MM. A review of therapeutic protein expression by mammalian cells. BioProcess International 2008; 6: 28-33
- Jiang Y., Li F., Zha D., Potgieter TI., Mitchell T., Moore R., Cukan M., Houston-Cummings NR., Nylen A., Drummond JE., McKelvey TW., d’Anjou M., Stadheim TA., Sethuraman N., Li H. Purification process development of a recombinant monoclonal antibody expressed in glycoengineered Pichia pastoris. Protein Expr Purif 2011; 76: 7-14
- Kim YK., Kim KR., Kang DG., Jang SY., Kim YH., Cha HJ. Expression of β-1,4-galactosyltransferase and suppression of β-N-acetylglucosaminidase to aid synthesis of complex N-glycans in insect Drosophila S2 cells. J Biotechnol 2011; 153: 145-152
- Chu L., Robinson D.K. Industrial choices for protein production by large-scale cell culture. Curr Opin Biotech 2001; 12: 180-187
- FDA: Comparability protocols - protein drug products and biological products - chemistry, manufacturing, and controls information. 2001
- Mack G. FDA balks at myozyme scale-up. Nat Biotech 2008; 26: 592
- Lipscomb ML., Palomares LA., Hernández V., Ramírez OT., Kompala DS. Effect of production method and gene amplification on the glycosylation pattern of a secreted reporter protein in CHO cells. Biotechnol Progr 2005; 21: 40-49
- Senger RS., Karim MN. Effect of shear stress on intrinsic CHO culture state and glycosylation of recombinant tissue-type plasminogen activator protein. Biotechnol Progr 2003; 19: 1199-1209
- Trummer E., Fauland K., Seidinger S., Schriebl K., Lattenmayer C., Kunert R., Vorauer-Uhl K., Weik R., Borth N., Katinger H., Müller D. Process parameter shifting: Part II. Biphasic cultivation—a tool for enhancing the volumetric productivity of batch processes using Epo-Fc expressing CHO cells. Biotech Bioeng 2006; 94: 1045-1052
- Yoon SK., Choi SL., Song JY., Lee GM. Effect of culture pH on erythropoietin production by chinese hamster ovary cells grown in suspension at 32.5 and 37.0°C. Biotechnol Bioeng 2005; 89: 345-356
- Borys MC., Linzer DIH., Papoutsakis ET. Ammonia affects the glycosylation patterns of recombinant mouse placental lactogen-i by chinese hamster ovary cells in a pH-dependent manner. Biotechnol Bioeng 1994; 43: 505-514
- Restelli V., Wang MD., Huzel N., Ethier M., Perreault H., Butler M. The effect of dissolved oxygen on the production and the glycosylation profile of recombinant human erythropoietin produced from CHO cells. Biotechnol Bioeng 2006; 94: 481-494
- Chee Furng Wong D., Tin Kam Wong K., Tang Goh L., Kiat Heng C., Gek Sim Yap M. Impact of dynamic online fed-batch strategies on metabolism, productivity and N-glycosylation quality in CHO cell cultures. Biotechnol Bioeng 2005; 89: 164-177
- Baker KN., Rendall MH., Hills AE., Hoare M., Freedman RB., James DC. Metabolic control of recombinant protein N-glycan processing in NS0 and CHO cells. Biotechnol Bioeng 2001; 73: 188-202
- Gu X., Wang DIC. Improvement of interferon-γ sialylation in chinese hamster ovary cell culture by feeding of N-acetylmannosamine. Biotechnol Bioeng 1998; 58: 642-648
- Crowell CK., Grampp GE., Rogers GN., Miller J., Scheinman RI. Amino acid and manganese supplementation modulates the glycosylation state of erythropoietin in a CHO culture system. Biotech Bioeng 2007; 96: 538-549
- Castro PM., Ison AP., Hayter PM., Bull AT. The macroheterogeneity of recombinant human interferon-gamma produced by chinese-hamster ovary cells is affected by the protein and lipid content of the culture medium. Biotechnol Appl Biochem 1995; 21 ( Pt 1): 87-100
- Borys MC., Dalal NG., Abu-Absi NR., Khattak SF., Jing Y., Xing Z., Li ZJ. Effects of culture conditions on N-glycolylneuraminic acid (Neu5Gc) content of a recombinant fusion protein produced in CHO cells. Biotechnol Bioeng 2010; 105: 1048-1057
- Mittermayr S., Bones J., Doherty M., Guttman A.s., Rudd PM. Multiplexed analytical glycomics: Rapid and confident IgG N-glycan structural elucidation. J Proteome Res 2011; 10: 3820-3829
- Gennaro LA., Salas-Solano O. On-line CE−LIF−MS technology for the direct characterization of N-linked glycans from therapeutic antibodies. Anal Chem 2008; 80: 3838-3845
- Marino K., Bones J., Kattla JJ., Rudd PM. A systematic approach to protein glycosylation analysis: A path through the maze. Nat Chem Biol 2010; 6: 713-723
- Siemiatkoski J., Lyubarskaya Y., Houde D., Tep S., Mhatre R. A comparison of three techniques for quantitative carbohydrate analysis used in characterization of therapeutic antibodies. Carboh Res 2006; 341: 410-419
- Campbell MP., Nguyen-Khuong T., Hayes CA., Flowers SA., Alagesan K., Kolarich D., Packer NH., Karlsson NG. Validation of the curation pipeline of unicarb-db: Building a global glycan reference MS/MS repository. BBA - Proteins and Proteom 2014; 1844: 108-116
- Campbell MP., Royle L., Radcliffe CM., Dwek RA., Rudd PM. GlycoBase and autoGU: Tools for HPLC-based glycan analysis. Bioinformatics 2008; 24: 1214-1216
- Stockmann H., Adamczyk B., Hayes J., Rudd PM. Automated, high-throughput IgG-antibody glycoprofiling platform. Anal Chem 2013; 85: 8841-8849
- Visser J., Feuerstein I., Stangler T., Schmiederer T., Fritsch C., Schiestl M. Physicochemical and functional comparability between the proposed biosimilar rituximab GP2013 and originator rituximab. BioDrugs 2013; 27: 495-507
- Berkowitz SA., Engen JR., Mazzeo JR., Jones GB. Analytical tools for characterizing biopharmaceuticals and the implications for biosimilars. Nat Rev Drug Discov 2012; 11: 527-540
- Engen JR. Analysis of protein conformation and dynamics by hydrogen/deuterium exchange MS. Anal Chem 2009; 81: 7870-7875
- Bennett CL., Luminari S., Nissenson AR., Tallman MS., Klinge SA., McWilliams N., McKoy JM., Kim B., Lyons EA., Trifilio SM., Raisch DW., Evens AM., Kuzel TM., Schumock GT., Belknap SM., Locatelli F., Rossert J., Casadevall N. Pure red-cell aplasia and epoetin therapy. N Engl J Med 2004; 351: 1403-1408
- Castelli G., Famularo A., Semino C., Machi AM., Ceci A., Cannella G., Melioli G. Detection of anti-erythropoietin antibodies in haemodialysis patients treated with recombinant human-erythropoietin. Pharmacol Res 2000; 41: 313-318
- Jiang H., Wu SL., Karger BL., Hancock WS. Characterization of the glycosylation occupancy and the active site in the follow-on protein therapeutic: TNK-tissue plasminogen activator. Anal Chem 2010; 82: 6154-6162
- Kawasaki N., Itoh S., Hashii N., Takakura D., Qin Y., Huang X., Yamaguchi T. The significance of glycosylation analysis in development of biopharmaceuticals. Biol Pharm Bull 2009; 32: 796-800
- Schiestl M., Stangler T., Torella C., Cepeljnik T., Toll H., Grau R. Acceptable changes in quality attributes of glycosylated biopharmaceuticals. Nat Biotech 2011; 29: 310-312
- Xie H., Chakraborty A., Ahn J., Yu YQ., Dakshinamoorthy DP., Gilar M., Chen W., Skilton SJ., Mazzeo JR. Rapid comparison of a candidate biosimilar to an innovator monoclonal antibody with advanced liquid chromatography and mass spectrometry technologies. MAbs 2010; 2
- Rathore AS., Winkle H. Quality by design for biopharmaceuticals. Nat Biotech 2009; 27: 26-34
- Ridgway A., Ritter N., Schiestl M., Schreitmueller T. Biosimilar products: Scientific principles, challenges, and opportunities. BioProcess Int 2013; 11: 12-20
- Harvey DJ., Merry AH., Royle L., P. Campbell M., Dwek RA., Rudd PM.: Proposal for a standard system for drawing structural diagrams of N- and O-linked carbohydrates and related compounds. Proteomics 2009; 9: 3796-3801
- ACTIP: Monoclonal antibodies approved by the EMA and FDA for therapeutic use. 2014. Available from: http://www.actip.org/pages/monoclonal_antibodiestable.html; [Accessed: 20.01.2014]

