


The European Medicines Agency
-
Copyright
© 2012 PRO MEDICINA Foundation, Published by PRO MEDICINA Foundation
User License
The journal provides published content under the terms of the Creative Commons 4.0 Attribution-International Non-Commercial Use (CC BY-NC 4.0) license.
Authors
| Name | Affiliation | |
|---|---|---|
Tadeusz Płusa |
Military Institute of Medicine in Warsaw, Department of Internal Diseases, Pneumonology and Allergology, Central Clinical Hospital of the Ministry of National Defence
|
The role and tasks of EMA
The Agency is responsible for the scientific evaluation of applications for European marketing authorisations for both human and veterinary medicines. Under the centralised procedure, companies submit a single marketing-authorisation application to the Agency.


All medicines for human and animal use derived from biotechnology and other high-tech processes must be approved via the centralised procedure. The same applies to all advanced-therapy medicines and human medicines intended for the treatment of HIV/AIDS, cancer, diabetes, neurodegenerative diseases, auto-immune and other immune dysfunctions, and viral diseases, as well as to all designated orphan medicines intended for the treatment of rare diseases. For medicines that do not fall under any of the above-mentioned categories, companies can submit an application for a centralised marketing authorisation to the Agency, provided the medicine constitutes a significant therapeutic, scientific or technical innovation, or is in any other respect in the interest of patient or animal health.
The Agency constantly monitors the safety of medicines through a pharmacovigilance network, and takes appropriate actions if adverse drug reaction reports suggest that the benefit-risk balance of a medicine has changed since it was authorised. For veterinary medicines, the Agency has the responsibility to establish safe limits for medicinal residues in food of animal origin.
The Agency also plays a role in stimulating innovation and research in the pharmaceutical sector. The Agency gives scientific advice and other assistance to companies for the development of new medicines. It publishes guidelines on quality-, safety- and efficacy-testing requirements.
Six scientific committees, composed of members of all EU and EEA-EFTA states, some including patients’ and doctors’ representatives, conduct the main scientific work. The Agency works with a network of over 4,500 European experts who serve as members of the Agency's scientific committees, working parties or scientific assessment teams. These experts are made available to the Agency by the national competent authorities of the EU and EFTA states.
The Agency can be considered as the 'hub' of a European medicines network comprising over 40 national competent authorities in 30 EU and EEA-EFTA countries, the European Commission, the European Parliament and a number of other decentralised EU agencies. The Agency works closely with its European partners to build the best possible regulatory system for medicines for Europe and protect the health of its citizens.
In view of the continuing globalisation of the pharmaceutical sector, the Agency works to forge close ties with partner organisations around the world, including the World Health Organization and the regulatory authorities of non-European nations. The Agency is continually involved in a wide range of cooperation activities with its international partners, designed to foster the timely exchange of regulatory and scientific expertise and development of best practices in the regulatory field.
The Agency is also involved in referral or arbitration procedures relating to medicines that are approved or under consideration by Member States in non-centralised authorisation procedures.
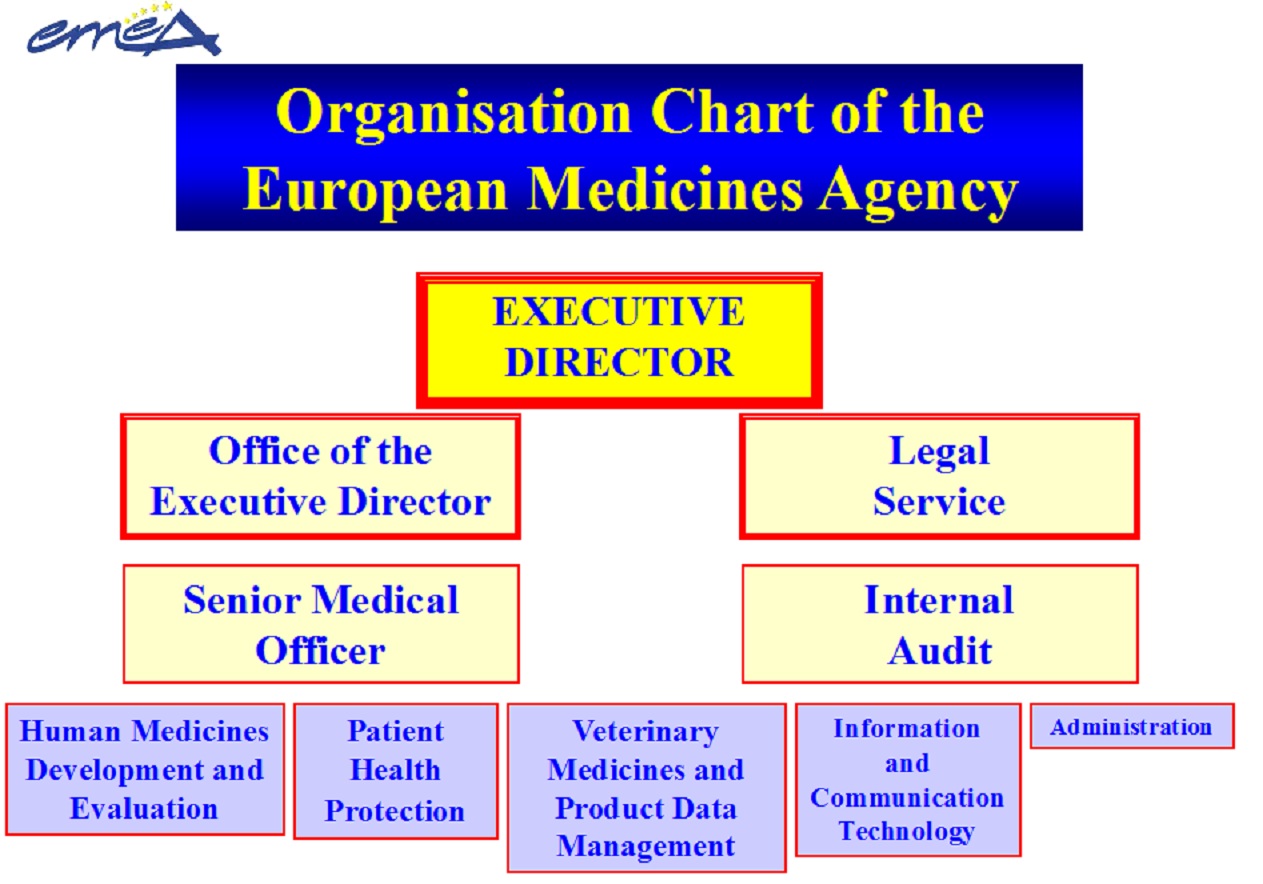
Structure of EMA
The organization cart of the European Medicines Agency is presented in figure 3.
The scientific evaluation of medicines in the European Union
The Agency is involved in the scientific evaluation of the hundreds of medicines that fall within the scope of the centralised procedure. However, thousands of other medicines that do not fall within this scope are marketed in the European Union either in individual Member States, in accordance with their national authorisation procedures, or in multiple Member States through the decentralised or mutual-recognition procedures. The Agency only becomes involved in the assesment of such medicines when they have been referred to the Agency due to a disagreement between two or more Member States about the authorisation or use of the medicine, or due to some other issue that requires resolution in the interest of protecting public health.
The Agency does not research or develop medicines, nor does it operate laboratories on its premises or elsewhere for such work. Research and development work on medicines is carried out by pharmaceutical companies or other medicines developers themselves, who then submit the findings and test results for their products to the Agency for evaluation.
Management Board
The Agency is governed by an independent Management Board. The Management Board consists of 35 members, who are appointed to act in the public interest and do not represent any government, organisation or sector. The Board sets the Agency’s budget, approves the annual work programme and is responsible for ensuring that the Agency works effectively and co-operates successfully with partner organisations across the EU and beyond. All Board members are required to make an annual declaration of any direct or indirect interests they have in the pharmaceutical industry. The Agency publishes these declarations of interest online.
The Management Board is an integral governance body of the Agency. It has a supervisory role with general responsibility for budgetary and planning matters, the appointment of the Executive Director and the monitoring of the Agency’s performance. The Board's operational tasks are very broad, ranging from adopting legally binding implementing rules, to setting strategic directions for scientific networks, to reporting on the use of European Union (EU) contributions for the Agency's activities:
It has legally enforceable rule-making authority for implementation of certain parts of the fee regulation, with the implementing rules being adopted and published as decisions of the Board. The Board also adopts the Agency's financial regulation and its implementing rules, which are binding texts for the Agency, the Board and the Executive Director.
It has a key role to play in the process whereby the EU budgetary authority gives discharge to the Executive Director for the Agency’s budget. The Board conducts an analysis and assessment of the Executive Director’s annual activity report, which is part of the package of controls and reports that lead to the discharge of the budget. The Board also gives its opinion on the Agency's annual accounts.
It has close ties with the Agency’s accounting officer, who is appointed by the Board, and with the internal auditor, who reports to the Board and to the Executive Director on audit findings. It is consulted on the rules of procedure of some of the Agency’s scientific committees, and on their membership. It is responsible for adopting the implementing provisions necessary for the practical application of the rules and regulations applicable to officials and other staff of the European Communities.
The members of the Management Board are appointed on the basis of their expertise in management and, if appropriate, experience in the field of human or veterinary medicines. They are selected to guarantee the highest levels of specialist qualifications, a broad spectrum of relevant expertise, and the broadest possible geographical spread within the EU.
The Management Board has 35 members:
- One representative of each of the 27 Member States;
- Two representatives of the European Commission;
- Two representatives of the European Parliament;
- Two representatives of patients’ organisations;
- One representative of doctors’ organisations;
- One representative of veterinarians’ organisations;
- In addition to the members, the Management Board also has one observer each from Iceland, Liechtenstein and Norway.
Executive Director

The Agency is headed by an Executive Director and has a secretariat of approximately 530 full-time staff. The Management Board is the supervisory body of the Agency, responsible, in particular, for budgetary matters. The Executive Director is the legal representative of the European Medicines Agency. He is responsible for all operational matters, staffing issues and drawing up the annual work programme.
The Agency's Executive Director is Guido Rosi (Photo 1.) who holds a degree in medicine and surgery, with specialisation in internal medicine, allergology and clinical immunology from the University of Rome. From 1978 to 1990, he worked as a physician in hospital, research and private practice. He worked from 1990 to 2008 in research at the Institute for Experimental Medicine of the National Research Council in Rome, directing the molecular medicine section from 2002 to 2005 and the Tor Vegata section from 2005 to 2008. He was made full professor of microbiology at the University of Rome “Tor Vegata” in 2008. From 2008 to 2011, Prof. Rasi was Director-General of the Italian Medicines Agency and a member of the European Medicines Agency’s Management Board. He joined the Agency as Executive Director on 16 November 2011.
The Agency's Deputy Executive Director is Andreas Pott, who also serves as the Agency's Head of Administration.
Agency ’s staff
The Agency’s staff is responsible for the administrative and procedural aspects of EU law related to the evaluation and safety-monitoring of medicines in the EU. All Agency staff are required to make an annual declaration of any direct or indirect interests they have in the pharmaceutical industry.
Scientific committees
The Agency’s six scientific committees are made up of independent professionals nominated by Member States from a pool of over 4,500 European experts. The committees are responsible for the scientific evaluation of marketing-authorisation application dossiers submitted by pharmaceutical companies, as well as for providing opinions on referrals and other issues impacting on public health, at the request of the Member States, the European Commission or the European Parliament. All committee members are required to make an annual declaration of any direct or indirect interests they have in the pharmaceutical industry. The Agency publishes these declarations of interest online.
Scientific evaluation on applications from pharmaceutical companies is carried out by six Scientific Committees (table 1). These Committees normally meet on a monthly basis and are comprised of members nominated by the Member States. Assessments are based on purely scientific criteria and determine whether or not the medicines concerned meet the necessary quality, safety and efficacy requirements (in accordance with EU legislation, particularly Directive 2001/83/EC). These processes ensure that medicines have a positive risk-benefit balance in favour of patients/users of these products once they reach the marketplace.
Table 1. Scientific Committees of EMA
| Committees of EMA | Abbreviations |
| Committee for Medical Products for Human Use | CHMP |
| Committee for Medicinal Products for Veterinary Use | CVMP |
| Committee for Orphan Medicinal Products | COMP |
| Committee for Herbal Medicinal Products | HMPC |
| Paediatric Committee | PDCO |
| Committee for Advanced Therapies | CAT |
The Agency’s scientific committees are not responsible for establishing ethical codes of conduct relating to the research or development of medicines, or for evaluating applications based on ethical considerations. Issues relating to ethics are established through legislation and directives set by the European Parliament, based on proposals from the European Commission, which in turn is advised by the European Group on Ethics in Science and New Technologies. As part of the initial administrative evaluation of the dossier of a marketing-authorisation application, the Agency’s role is to ensure that previously determined standards and regulations have been implemented.
The Agency works with a number of international organisations and standardisation initiatives (figure 4 and 5).


Agency cooperates in Poland with the Main Pharmaceutical Inspectorate and the Office for Registration of Medical Products, Medical Devices and Biocidal Products. Our opinions and scientific evaluations are presented in Scientific Committees in all fields of EMA activities.

